Compliance
The primary objective of the SF State Compliance Program is to provide information, training, and internal controls that are needed to meet the laws, rules, and policies governing sponsored research at SF State. The Compliance Committee, chaired by the Provost and Vice President for Academic Affairs, oversees the Compliance Program. The Compliance Team manages the daily operation of the sponsored research Compliance Program. The Compliance Team's chief responsibility is to monitor internal controls to ensure compliance with all the applicable federal laws and regulations that regulate sponsored research funding.
Please contact Jenny Chau, Compliance Officer (415-338-1862, jchau@sfsu.edu) if you have any questions about the SF State Compliance Program. The following areas represent the subcategories of SF State's Compliance Program: Responsible Conduct of Research, Conflict of Interest, Time and Effort Reporting, Cost Sharing, Sub-Recipient Monitoring.
For more information about Compliance
Human and Animal Protection
Human and Animal Protections at San Francisco State University supports the work of the Institutional Review Board (IRB) and the Institutional Animal Care and Use Committee (IACUC).
The IRB and the IACUC are charged with protecting the safety and welfare of humans and animals used in research at or in conjunction with this university. The committees do not expect research to be free from risk, but do expect the investigator to be aware of the risks, to minimize risk when possible, and to take appropriate precautions whenever necessary.
Phone: (415) 338-1093
Email: protocol@sfsu.edu
HAP OFFICE HOURS are On-Demand Monday-Friday email protocol@sfsu.edu to set up a specific day/time.
More information about SF State Human and Animal Protection: Human Subjects Research (IRB) or SF State Human and Animal Protection: Animal Subjects Research (IACUC).
2021-2022 Meeting Dates:
February 2, 2022
March 2, 2022
April 6, 2022
May 4, 2022
Helpful IRB Manger Tip:
If you click on the Start ADE link above or Click here to submit an ADE Form/IRB Protocol or any form in the Start xForms in IRB Manager, a new form opens; this form automatically saves if you close the tab or click the "Save for Later" button.
To find that form again look under the "xForms section of your dashboard, You have # of un-submitted xForms."
SF State IRB Guidance for Human Subject Research during COVID-19
Announcing Three (3) Year Approval for Qualifying Minimal Risk Research
Utilizing flexibility available under our Federalwide Assurance (FWA) regarding certain study approval periods, the Institutional Review Board and ORSP - Human and Animal Protections are pleased to announce the following change in policy. Effective May 1, 2014, the IRB will begin issuing three (3) year approvals for faculty research that qualifies for this extended approval period for new protocols. To qualify, the research must:
- involve no more than minimal risk to participants (as defined by Federal Regulations for Human Dubjects Research);
- not be supported by federal funds; and
- not be subject to federal oversight.
To extend the expiration date on existing approved protocols, please contact our office directly for more information.
Start the Application for Determination of Exemption -or- Go to IRB Manager Home Page
Please note that ADE submissions require a completed CITI training certificate entitled "Social/Behavioral Research Course."
Online CITI Research Training
All individuals conducting human subjects research are required to pass an online research training course. This includes not only the principal investigator but also all co-investigators, research technicians, research assistants, or student assistants who have contact with the research participants or identifiable data.
A course completion report will be issued upon completion of either course. The report must be submitted with the protocol materials. The reports are valid for 3 years from the date of issue. Certificates that expire before the 1-year protocol approval period will not be accepted for review.
The CITI course at Citi Program, Research, Ethics and Compliance Training Website is recommended for social/behavioral/educational researchers because of the elective modules which may be tailored to your own field of study.
CITI is free for SF State students, faculty, and staff.
For first time users, please select "Register" at the CITI homepage and on the following screen, enter "San Francisco State University" for the Organization Affiliation. Please select Social/Behavioral Research Course or Basic Human Subjects - Social/Behavioral Focus. Please note: Responsible Conduct of Research: Social and Behavioral Course does not fulfill this requirement.
For those who have protocols approved with NIH trainings in the past, you will need to update your human subjects research trainings through CITI program by August 1, 2019. The IRB requires current (not expired) completed human subjects training certificates for IRB approvals.
Please contact us if you have any issues with our online form or questions!
Logging into IRB Manager
ADE Application Submission Part 1
ADE Application Submission Part 2
ADE Application Submission Part 3
ADE Application Submission Part 4
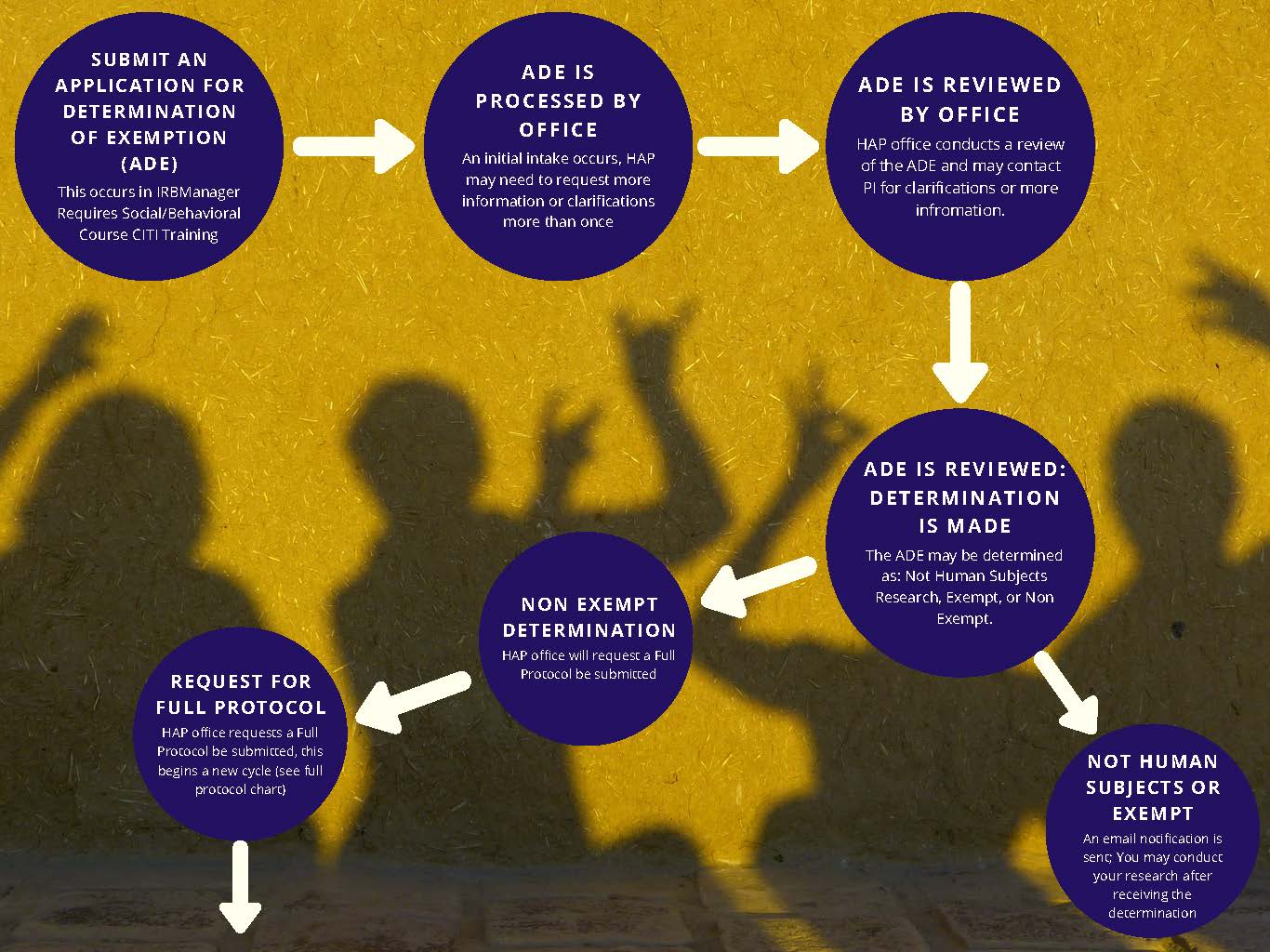
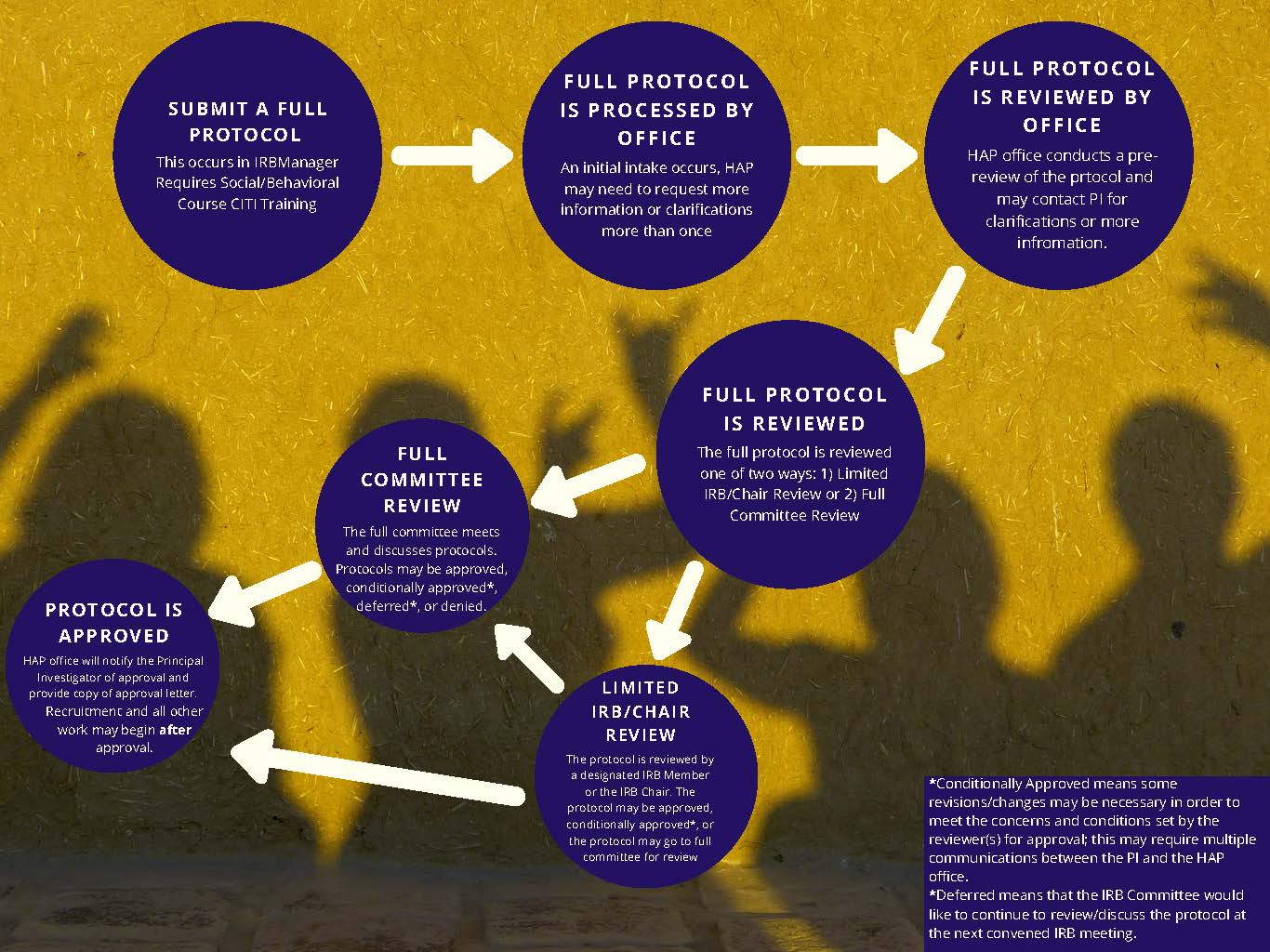
Caption of IRB Review Cycles images:
- Submit an Application for Determination of Exemption (ADE)—this occurs in IRB Manager and requires “Social/Behavioral Course” CITI Training.
- ADE is Processed by Office—an initial intake occurs, HAP office may need to request more information or clarifications more than once
- ADE is Reviewed by Office—HAP office conducts a review of the ADE and may contact the PI for clarifications or more information
- ADE is Reviewed and a Determination is Made—the ADE may be determined as Not Human Subjects Research, Exempt, or Non Exempt.
- Not Human Subjects Research or Exempt Determination—an email notification is sent via IRB Manager; you may conduct your research after receiving the determination.
- Non Exempt Determination—HAP office will request a Full Protocol be submitted via an email from IRB Manager. The Full Protocol is also submitted via IRB Manager. Proceed to Step 5.
- Request for Full Protocol—HAP office requests a Full Protocol (called an IRB Protocol Submission), this begins a new cycle
- Submit a Full Protocol—this occurs in IRB Manager use xForm: IRB Protocol Submission, requires “Social/Behavioral Research Course” CITI Training.
- Full Protocol is Processed by Office—an intake occurs, HAP may need to request more information or clarifications more than once
- Full Protocol is Reviewed by Office—a full protocol is reviewed one of two ways either limited IRB review/Chair Review or Full Committee Review
- If Full Committee Review—the full IRB Committee meets and discuss protocols. Protocols may be approved, conditionally approved*, deferred*, or denied.
- If Limited IRB/Chair Review—the full protocol is reviewed by a designated IRB member or the IRB Chair. The protocol may be approved, conditionally approved*, or the protocol may go to full IRB Committee for review.
- Protocol is Approved—HAP office will notify the Primary Investigator of approval and provide an approval letter and stamped consent forms (if applicable) via an email from IRB Manager. Recruitment and all other work may begin after approval.
Note: Conditionally Approved means some revisions/changes are necessary in order to meet the concerns and conditions set by the reviewer(s) for approval. This may require multiple communications between the PI and the HAP office. Deferred means that the IRB Committee would like to continue to review/discuss the protocol at the next convened IRB meeting.
- Informed Consent
- Parental Permission
- Minor Assent to Participate in Research
- Implied Consent for Surveys
- Verbal Consent Form
- Witness Translator Consent Form
- Focus Group Consent Form
- Permission to Recruit / Conduct Research
- Recruiting Script
- Photo Release
- Video Release
Utilizing flexibility available under our Federalwide Assurance (FWA) regarding certain study approval periods, the Institutional Review Board and ORSP - Human and Animal Protections are pleased to announce the following change in policy. Effective May 1, 2014, the IRB will begin issuing three (3) year approvals for faculty research that qualifies for this extended approval period for new protocols. To qualify, the research must:
- involve no more than minimal risk to participants (as defined by Federal Regulations for Human Subjects Research);
- not be supported by federal funds; and
- not be subject to federal oversight.
To extend the expiration date on existing approved protocols, please contact our office directly for more information.
More information about SF State IRB
The Institutional Review Board (IRB) for San Francisco State University is charged with reviewing all research involving research volunteers to ensure that their rights are protected according to the federal regulations stated in 45 Code of Federal Regulations, Part 46 and 21 CFR Parts 50 and 56 (FDA policy).
The IRB at SF State is composed of individuals from a number of units on campus, along with community members who are not affiliated with the university. IRB members are appointed by the Associate Vice President for Research and Sponsored Programs and the Dean of Graduate Studies with the concurrence of the Executive Committee of the Academic Senate.
The IRB generally meets on the first Wednesday of each month during the fall and spring semesters. They also meet intermittently during the winter and summer sessions, as needed. For protocols that reach full committee review and are non-exempt, the turnaround time is four to eight weeks.
Most of the protocols that the IRB reviews fall into the social and behavioral sciences category. The IRB include a number of qualitative researchers, as well as non-scientists and those engaged in quantitative research. Community members are included to provide a non-SF State perspective on university research. The IRB cannot meet without at least one non-scientist present.
Here is the current SF State Academic Senate Policy regarding use of Human Subjects in Research at SF State.
2021-2022 Meeting Dates:
September 1, 2021
October 6, 2021
November 3, 2021
December 1, 2021
February 2, 2022
March 2, 2022
April 6, 2022
May 4, 2022
Protocol Review Process
Once your complete IRB Protocol is received, we will review your documents to determine the category of review which your protocol will undergo and conduct a pre-review for needed revisions. We may request more information.
If the category is determined to be Exempt, no further review is needed and you will receive an Exemption notice in the form of an email from protocol@sfsu.edu. You may begin your research upon receiving the Exemption notice, but you must notify us of any changes in your study. Please note that you and your faculty advisor are responsible for ensuring all documents are free of grammatical and typographical errors.
If the category is determined to be Expedited, we will work with you to prepare and revise your protocol for a designee of the IRB, which is currently the IRB Chair. The IRB Chair will review the protocol and may request additional changes or approve the protocol. Once approved, we will send an approval letter via email from protocol@sfsu.edu. You may begin your research upon receiving the approval letter, but you must notify us of any changes in your study and renew your project one year from the date of approval.
If the category is determined to be Full Committee (also known as non-Exempt), we will work with you to prepare and revise your protocol for the Institutional Review Board (IRB). After a pre-review and requested revisions are addressed, the protocol is sent to the IRB members and reviewed at the next available IRB meeting. The IRB will issue a letter within 7-10 days after the meeting with their decision. The IRB may:
- Approve your protocol
- Defer your protocol because substantial changes are needed and changes must be reviewed by the IRB at a future meeting.
- Grant contingent approval - this means that you may not begin your research until you address a list of revisions as requested by the IRB. Once the contingencies (revisions) to your protocol are submitted and reviewed by the IRB Chair, we will send an approval letter from protocol@sfsu.edu. You may begin your research upon receiving the approval letter, but you must notify us of any changes in your study and renew your project one year from the date your protocol went to the IRB meeting.
Expected Review Turnaround Time
While the rate of the review process greatly depends on the queue of protocols preceding the researcher's submission and the researcher's promptness in responding to revision and clarification requests, approximate review time frames are as follows:
Exempt: 1-2 weeks
Expedited: 2-4 weeks
Non-Exempt (Full Committee): 4-8 weeks
During the fall and spring Graduate Studies deadline periods are as follows:
Exempt: 2-3 weeks
Expedited: 3-6 weeks
Non-Exempt (Full Committee): 4-8 weeks
More information about SF State IACUC
An animal protocol must be submitted to HAP to begin the review process for any research and teaching projects using live, vertebrate, non-human animals, as required by federal law and SF State policy.
In addition, all researchers using animal subjects are required to complete the online Animal Welfare course, "Working with the SF State IACUC," which takes approximately 2 hours. A course completion report will be issued after you complete the course. The certificate must be provided to HAP. The certificate is valid for 3 years. Protocols cannot be approved without this certification.
To register for the course, go to CITI Program, Research, Ethics, and Compliance Training Website
- Proceed directly to "New Users Register Here."
- Choose "San Francisco State University" from the drop-down menu.
- Click "Submit."
- Choose a user name and password. Write them down. You will need them again if you choose to take the course in more than one session, or if you ever need another copy of your course completion report.
- Fill out the required fields in the next two screens. When directed to the CITI course enrollment procedure page, please scroll down to the bottom of the page and click on "Question 1."
- The required course is "Working with the SF State IACUC." When you finish the required course, optional courses will be available.
- Scroll down to the bottom of the page and click on "Submit." You will be redirected to the Learner's Menu.
- Scroll halfway down the page and click on "Not Started-Enter."
This will take you to the Introduction and modules required to complete the course.
To submit an animal protocol click here
IACUC policy on visitors to the Animal Quarters
All requests for access to the animal quarters for the purpose of bringing visitors must be made at least two weeks in advance. These requests must be sent to the IACUC Chair and Committee for review prior to the visitor's arrival.
- Q1: I am conducting a secondary analysis of existing data. Do I need to obtain approval from the IRB?
A: Yes. Research involving the secondary analysis of existing data must be reviewed by the IRB to ensure that the original data were properly and ethically obtained and that the objectives of the secondary analysis are in keeping with those for which consent was obtained.
- Q2: I think my research is exempt. Do I still need to hand in a protocol?
A: Yes. All research that involves research volunteers, exempt and non-exempt, must be reviewed by the Institutional Review Board (IRB) or by the IRB office.Check our section on "Does My Project Require Review?" to find out if your research does not involve research volunteers, or does not fit the definition of "systematic research designed to develop or contribute to generalizable knowledge."
- Q3: Why do I have to get letters of permission?
A: A person in authority must be aware of and approve your recruiting from among the clients, students, etc. of the organization through which you plan to recruit participants or conduct your research. Please note that if you intend to conduct research on the same premises, or with the same group, the letter should also include permission to conduct research with this population, as well as recruit volunteers from it. This is especially true if you will be using class time, client appointment time, etc. to administer your survey or conduct your interviews.The person signing the permission letter must also know what your research topic and title are. Frequently it is easier for you to write the letter for them, have their office copy it on school or agency letterhead, and then have the director or principal/assistant principal sign it. We recommend you use our Permission Letter template.
- Q4: I am interviewing Key Informants for my research. Do they need consent forms?
A: Key informants or content experts or a panel of experts do not require consent forms, because they are not considered research volunteers needing protection if you are asking them questions that relate solely to their field of expertise, and the questions are factual in nature. However, as a courtesy, you should provide them with a brief information sheet with your contact information, your advisor's contact information and the title of your research.
- Q5: Why do I need a literature review in my protocol statement?
A: Federal regulations regarding ethical research require that risks to research volunteers be minimized by using procedures which are consistent with sound research design, and which do not unnecessarily expose volunteers to risk (45 CFR 46.111(a)(1).A literature review:
allows the IRB to know more about your topic, demonstrates that you are aware of prior research in this field, including hypotheses, methods, and outcomes of previous research, indicates that this knowledge has informed your proposed use of research volunteers, should justify and support the purpose of your own study and its importance in your academic field. - Q6: Why do I need to include my data analysis methods?
A: A discussion of your data analysis methods indicates that you have a plan to evaluate your data in the light of your research question. If your data will not answer your research question or hypothesis, your study will have no benefit and even the slightest risk will be outweighed by the lack of benefit. In this case you will need to revise your protocol, instruments, and/or analysis methods to make sure you will elicit the data that will answer your research question or fulfill the research purpose.The data analysis section should address all sources of data you are collecting. Describe the method of analysis, any software you will use, any collection notes or data sheets you have devised to help you collect information, etc.
If you are conducting a qualitative study, state specifically which qualitative method of analysis you will use, cite the relevant literature, and tell us what that means with regard to your own research. For instance, if you are collecting interview data and looking for common themes throughout, please state this, mention potential themes, and reference Glaser and Strauss in your lit review. If you are using the data anecdotally to support your own observations, field notes, or journals, please state this.
- Q7: Why do I need to include the age of the research volunteers?
A: The IRB uses age of research volunteers as one of the criteria used to determine whether your research is exempt or non-exempt. Volunteers under 18 years old are minors, and considered a vulnerable population. Research with minors would make an otherwise exempt survey or interview non-exempt, and would call for a full committee review, which can add time to the approval process. Interviewing or surveying adults is almost always exempt. - Q8: What does "identifiable information" mean?
A: Identifiable information is any information that can single out a research volunteer. This includes, but is not limited to: name, job title, age, fingerprints, biometric data, gender, birth date, ethnicity or race, medical records number, student ID number, and even zip code if less than 20,000 people live in that zip code. Please tell us what information on your volunteers you do have or will collect, and we can then determine if it is identifiable for your research population.. "Unidentifiable" is not the same as anonymous, which only refers to not knowing someone's name. While you may not know or use a research volunteer's name , you may still be able to identify that volunteer through a combination of other identifiers. - Q9: What should I include in my "recruiting script"?
A: "Recruiting scripts" refer to the content of an in-class presentation or a telephone call, the text of an e-mail, letter, or newspaper advertisement, or the copy of a flyer you will post to inform prospective volunteers about your research. These scripts should be brief ( about a paragraph long) and objective. They should be a simple invitation to participate in the research.Recruiting scripts should briefly include: who you are, what your study is about, what will happen to the volunteers, when and where it will happen, and how long it will take, and your contact information. Include specific inclusion or exclusion criteria for participation on your script (must be 21, must be computer science students, must have asthma, etc.).
- Q10: What happens after I email my protocol to protocol@sfsu.edu?
A: First, a reviewer determines its status and screens the complete protocol package to ensure that all necessary documents are included. The review process does not begin until all documents are submitted.Exempt protocols will be certified as such in the office. You will receive an e-mail notifying you of its exemption from further review.
Next, your protocol enters the pre-review phase. A protocol analyst looks for substantive issues that need clarification or revision to bring the protocol into compliance. At this point, an email will be sent to the researcher, requesting changes. Once revisions are received, follow-up requests may occur.
We communicate exclusively by e-mail, so all requests for changes, clarifications, or additions will be sent from protocol@sfsu.edu to the email address provided on your Protocol Approval Form.
Please check with the office if you haven't heard that your protocol or revisions have been received. We send the e-mail confirmation within a week of a protocol's arrival.
After the November 1 deadline for the next spring semester's thesis courses, the office is swamped with protocols. Be aware that at this time, protocols go through your department for signature, then through the Grad Studies office, then come to our office, so there will a time lag between your turning your protocol in to your department and our office receiving it.
- Q11: What happens to my revisions?
A: Once your revisions are submitted, they will be reviewed in the order in which they are received, to make sure all requested changes have been made, and to review any additional material you may have provided.More revisions may be requested to correct any errors or omissions in the newly-submitted material, to include something that was missed the first time around, or to request again any prior changes that haven't been made.
Non-exempt protocols will go to the committee for a full review, once all revisions have been submitted by the researcher(s). In the period right after the semester deadlines, the committee reviews the bulk of the protocols we receive all year, so it may take some time to get on the agenda for a meeting. You will receive an e-mail once your protocol has been placed on an agenda.
The IRB will very likely require more changes after its review. You will receive an email from the IRB chair, notifying you of your protocol?s review status, and any final changes required by the committee.
Non-exempt protocols will receive an official letter of approval with the protocol number, date of approval, and an expiration date. Date of approval will be the date of the full committee review, regardless of when your final changes were accepted and approved. The letter will be in PDF format and attached to an e-mail message.
- Q12: What are the options after full committee review?
A: The IRB can give full approval, contingent approval (contingent on fulfilling some minor conditions before full approval is granted), or can defer your protocol for serious substantive changes. If the protocol is deferred, the revisions will have to return to the committee for approval. If given contingent approval, the revisions can be approved in the office and signed by one of the committee chairs. You have not been fully approved until you receive the official letter of approval from the IRB office.
- Q13: When is the date of approval?
A: If your protocol is approved immediately, the approval date will be the date of the full IRB meeting at which the protocol was reviewed. If it's given contingent approval, the approval date will also be the date of the full IRB meeting, regardless of when your final revisions are received and approved. If it's deferred, and your revisions are reviewed at the second IRB meeting and accepted, the approval date will be the date of the second full IRB meeting.
- Q14: How long does the approval last?
A: All protocols have to be reviewed once a year, at a minimum. Protocol approvals expire one year from the date of the full IRB meeting at which the protocol was approved or contingently approved.However, the IRB may approve protocols for less than a year if they feel the risks, population, consent issues, or other considerations warrant an earlier review.
- Q15: How long does it take to get approval?
A: The process usually takes between 4-12 weeks. This timeline is contingent upon receipt of a full protocol package and quick responses from the researcher. During our "high season," that is, between November 1 and February 1, when we receive the bulk of protocols from spring graduates, review can take longer due to full IRB agendas.For this reason, we encourage students to consider exempt research projects, because they do not require a full IRB review. They can be reviewed and certified as exempt from further review by administrative staff in the office.
- Q16: How do I make changes to a protocol that has already been approved?
A: Submit a Modification Request form. Explain the changes you want to make and your reasons for making them. Attach all revised IRB documents. For instance, if you are changing the recruiting information, submit the revised flyer, recruiting script or text. Highlight changes so we can compare the old and new documents. If the changes are substantive, or involve higher risk to the research volunteers, you would also have to incorporate the changes in a revised protocol, which may have to go to the full IRB review.
- Q17: Once my protocol is approved, does anyone ever check to see I am following it?
A: Student advisors have let us know in the past if they were afraid a student's research was going off in an unapproved direction, or hadn't been explained fully, and we have worked with the student to correct the errors, or incorporate the modification as approved.One of the IRB's responsibilities is to monitor research, especially the recruiting and consent processes and documents.
- Q18: How can I get help on the application process?
A: Do explore this web site. All the information, forms and templates are here. If you still need help, contact our office via email at protocol@sfsu.edu or by telephone at (415)338-1093 . We can answer any questions you may have about the protocol process. We are also available to meet with you by appointment to discuss your draft protocol, draft consent or draft culminating experience description.
- Q19: What should I know about using deception in my research?
A: While exploring your area of interest may require misleading or not completely informing your research volunteers about the true nature of your research, as a general rule, serious deception should be avoided whenever possible, since it jeopardizes the integrity of informed consent. Federal regulations prohibit the use of deceptive techniques that place research volunteers at greater than minimal risk. Human and Animal Protections will review any protocol that uses deception very closely. Deception includes, but is not limited to:
Intentionally misleading participants about their status
Giving false information about the investigators or the research purpose
Omitting information about the real purpose of the researchFor research involving deception:
the use of deception must be justified in the protocol to show that the research cannot be performed in the absence of deception and the benefits of the research will sufficiently outweigh any risks that deception may create; research participants cannot be deceived about significant aspects of the research that would affect their willingness to participate or that would cause them physical or emotional harm; and deception must be explained to participants (debriefed) as early as feasible. A debriefing script must be included in the protocol and should include a detailed description of the ways in which deception was used and why; when and by whom the debriefing will be administered should also be included; and true "informed consent" cannot be given if the true nature of the research is deceptively presented. This situation is dealt with administratively. Research employing deception may not be reviewed as "Exempt." Deceptive research that involves mild deception [as determined by Human and Animal Protections or the Institutional Review Board (IRB)] or omission (e.g., participants not of the true purpose of the research) where the topic is not sensitive and the research volunteers are not vulnerable can be reviewed as "Expedited." All other deceptive research will be reviewed as "Non-exempt with Full Committee Review."
The IRB may suggest that the investigator add a sentence to the consent form such as "Research designs often require that the full intent of the study not be explained prior to participation. Although we have described the general nature of the tasks that you will be asked to perform, the full intent of the study will not be explained to you until after the completion of the study." Investigators may be asked to include an option for participants to withdraw their data from the study after they learn the true nature of the research, if it is of a sensitive nature.
- Q20: What is the policy on dietary supplements?
FDA definition from Dietary Supplement Health and Education Act (1995): composed of essential nutrients, such as vitamins, minerals, and proteins, herbs or similar nutritional substances (such substances as ginseng, garlic, fish oils, psyllium, enzymes, glandulars, and mixtures of these).It is intended to supplement the diet and bears or contains one or more of the following ingredients: a vitamin; a mineral; an herb or other botanical (excluding tobacco); an amino acid; a dietary substance for use by humans to supplement the diet by increasing the total daily intake (e.g., enzymes or tissues from organs or glands); a concentrate, such as energy bars; or a metabolite, constituent, or extract.
It is intended for ingestion in pill, capsule, tablet, food substance, or liquid form, or administered by another route (e.g., patch).
It is not represented for use as a conventional food or as the sole item of a meal or diet.
It is labeled as a "dietary supplement".FDA regulates dietary supplements as foods, and not as drugs.
Dietary Supplement Health and Education Act
National Institutes of Health Office of Dietary Supplements
Disclaimer: The information provided by the investigator to research volunteers is for informational purposes and should not be construed as medical advice. Volunteers should consult with a qualified health care professional for advice about their specific health or medical needs before taking any dietary supplement.
Introduction
Review of dietary supplement research will be on a case-by-case basis, based upon valid reports in the existing, established and mainstream literature of risks encountered with the formulation and suggested dose of the supplement, the population proposed for the research, and the actual research procedures. For supplements and/or a subject population and/or a procedure deemed to be higher risk, the review will be more stringent and the committee may require consultation with or oversight or co-investigation by a health care professional. The committee reserves the right to prohibit research deemed too risky, either because of the supplement itself, or the population to be studied, or the actual procedures or any combination.Policy
Safety of the supplement and its proposed use (population and dose)
As well as the existing requirement for a brief (1 - 2 paragraphs), current, scholarly review of relevant literature that supports the purpose of the research study, the researcher must include a review of the clinical research detailing the presence/absence of the side effects associated with use of this supplement, particularly when administered to a population and in a dose and duration similar to those which the researcher is using. This should include both minor and major side effects, and frequency. Inclusion of brand-names may be relevant if some brands have been associated with side effects, but not others.
The committee can request a validation of the literature by either Human and Animal Protections, if appropriate personnel are available, or by an outside consultant.
All possible side effects should be stated in lay terms [e.g., hypotension (low blood pressure], and frequency of occurrence included, especially for serious side effects.
A signed, adequate health history/screening questionnaire will be required to reduce the risk to volunteers by screening out potential high risk volunteers.
Certain medications being taken by a potential volunteer may result in their exclusion from the study. These medications should be pre-determined by the researcher by a thorough review of the relevant clinical research literature and they should be specifically listed on the health history/screening questionnaire.
The researcher should demonstrate an understanding and knowledge of the side effects and how serious they are.
Volunteers should be told to seek medical or emergency treatment for side effects, as appropriate, as well as notifying the researcher. Obviously, things such as severe headaches, fainting or extensive bleeding require immediate medical attention.
Volunteers should be advised that these supplements may interfere with other supplements or medications they are taking. Also, they should be advised to tell their physician that they are taking/have taken this supplement when they meet with him/her.
The researcher must stay abreast of current literature and studies and notify the Institutional Review Board (IRB) immediately if any additional risks are identified for this supplement. The committee will then determine if the study needs to be altered or terminated, or if the consent documents need to be changed to reflect these additional risks.
Study design
Although the committee usually does not require changes to a researcher's study design, it does have the prerogative to do so when the design contributes to the risk encountered by the subject. For dietary supplement research, the committee may request changes in the design to either (1) reduce the risk to the volunteer or (2) increase the benefit of the research to justify additional risk.
To avoid any appearance that the researcher may be seen to be "dispensing" supplements, we have adopted a procedure similar to that used at the University of California, San Francisco. This procedure involves having the supplements and the placebos and their directions out on the table, and potential volunteers are told they may choose to take the supplements, if they wish.
Disclaimers should be included on consent forms for (1) the fact that the supplement has not been approved by the FDA and (2) "this study is not affiliated with a medical center treatment or trial (and drug or supplement company, if appropriate.
Any personnel associated with the study should not be identified as "Dr." unless they are a medical doctor to avoid confusion on the part of a volunteer.
Research Volunteers
Research with vulnerable or patient populations would require more stringent review by the committee than research on normal volunteers. The committee may require additional literature review that addresses this population. The committee may require consultation with or oversight or co-investigation by a medical health professional.
Health questionnaire/screen
The health history/screening questionnaire is paramount in reducing risk to the research volunteers. Whenever possible, all volunteers with any pre-existing condition or symptom that may put them at risk when taking this supplement should be excluded. If this is not possible (e.g., the need for a special category of subjects), the committee may require a health care professional to oversee the medical history, provide clearance for the volunteer's participation, etc. The cost of this oversight would be the responsibility of the researcher.
If the risks associated with the supplement are minor or rare, then a signed, self questionnaire should be adequate. If the risks are of more concern, either because of the supplement or the population, we may require a health care professional to oversee the medical history or provide clearance for the volunteer's participation. The cost of this oversight would be the responsibility of the researcher.
Supplement itself
Is the supplement routinely available to the public over-the-counter? Research where the supplement has been made available to the researcher specifically for this project may require more stringent review, if there is conflict of interest. If the production of this product is under the quality control of, e.g., a pharmaceutical company, this may lessen the stringency of review.
Volunteers should be told what the role of the dietary supplement supplier is in the research-uninvolved (in which case, the researchers would be providing the supplement themselves), providing the supplement as a courtesy/convenience to the researcher, or sponsoring the research.
Details of the placebo also need to be included.
While the IRB recognizes that the supplement industry does not always conduct expensive controlled trials and may instead rely upon "testimonials", research on supplements that have not undergone controlled clinical testing may not be able to be approved by the IRB. Evidence documenting the effects of individual components of the supplement may not be acceptable unless clinical data about their interaction with the other components in the product is available
- Q21: Where can I find more information about certificates of confidentiality?
For more information and policies regarding Certificates of Confidentiality, please visit the following National Institute of Health website:
http://grants.nih.gov/grants/policy/coc/
- Q22: What are the turnaround times for submitted protocols?
A: While the rate of the review process greatly depends on the queue of protocols preceding the researcher's submission and the researcher's promptness in responding to revision and clarification requests, approximate review time frames during the fall and spring Graduate Studies deadline periods are as follows:Exempt: 1-2 weeks
Expedited: 3-6 weeks
Non-Exempt (Full Committee): 4-8 weeksResearch with research volunteers - including recruitment of research volunteers - cannot proceed until the researcher receives final approval from the protocol office via e-mail from protocol@sfsu.edu. Proceeding with any part of the research or using data collected before final approval can result in suspension from the university.
- Q23: What should I consider when using a raffle prize as an incentive?
A: The value of the prize or the amount of money given should not be coercive. That is, participants should not be likely to fill out the survey only for a chance to win. Researchers should consider their target population and the average socioeconomic status of the participants when determining the prize amount/value.The following information should be provided in your Informed or Implied Consent:
"Participants are eligible to enter a raffle for their contribution to this research. The prize is a $$$ giftcard/gift certificate/e-voucher/e-giftcard/product/item and the odds of winning will depend on the total number of entries received. The raffle drawing will take place on xx/xx/xx and the researcher/advisor/third party is responsible for drawing the winner. Participants will be eligible to enter the raffle even if they do not complete the survey. Please contact the researcher for more information regarding the raffle and/or click through to the end of the survey to enter the raffle/prize drawing."
- Q24: How can I streamline the review and revision process?
A: When submitting:
Consider conducting an exempt research project, which will not need full committee review and will not expire. We can advise an investigator prior to submission whether or not a project will qualify for exemption. In general, surveys and interviews of adults, about non-sensitive topics, are considered exempt.See Review Categories: Research that does not need review to determine whether the research even requires IRB review. While the committee's charge is broad, there are several categories of projects that are not considered "research" in the NIH sense, or are research but do not involve research volunteers.
Submit all documents before the semester deadline if you plan to graduate in the following semester. Please be aware that each department and college has its own, usually earlier, deadlines that the student must follow. Generally, documents are submitted to the department office for the final signatures by graduate coordinator or department chair, then forwarded to the IRB office by the department.
Submit a complete protocol package from the start.
State your research purpose or question clearly. Know what research has been done in the field recently, and how that affects your study. Are you trying to replicate earlier results, or are you trying to prove something different?
Explain your procedures clearly and concisely. Include the details. Tell the IRB and the volunteers when, where, and how long the research will be, and exactly what they will experience during the research procedures. Tell them what you would like to know before volunteering as a research volunteer. This is the meaning of "informed consent."
When revising:
- The single most frequent cause of delays in protocol approval is the time lag between the office's request for revisions, and the researcher's reply to that request.
- Return revisions in a timely manner, because they are reviewed in the order they are received.
- Make all the changes requested, or explain why a requested change was not made. There may be a very good reason for not making a change, due to the research purpose or methods, an advisor's recommendation, or the requirements of the discipline. State what these reasons are.
- With all revisions, submit a cover letter explaining all changes, including any that were not requested by our office. Highlight all changes for faster review.